La ANMAT autorizó el uso de emergencia de la vacuna de Janssen
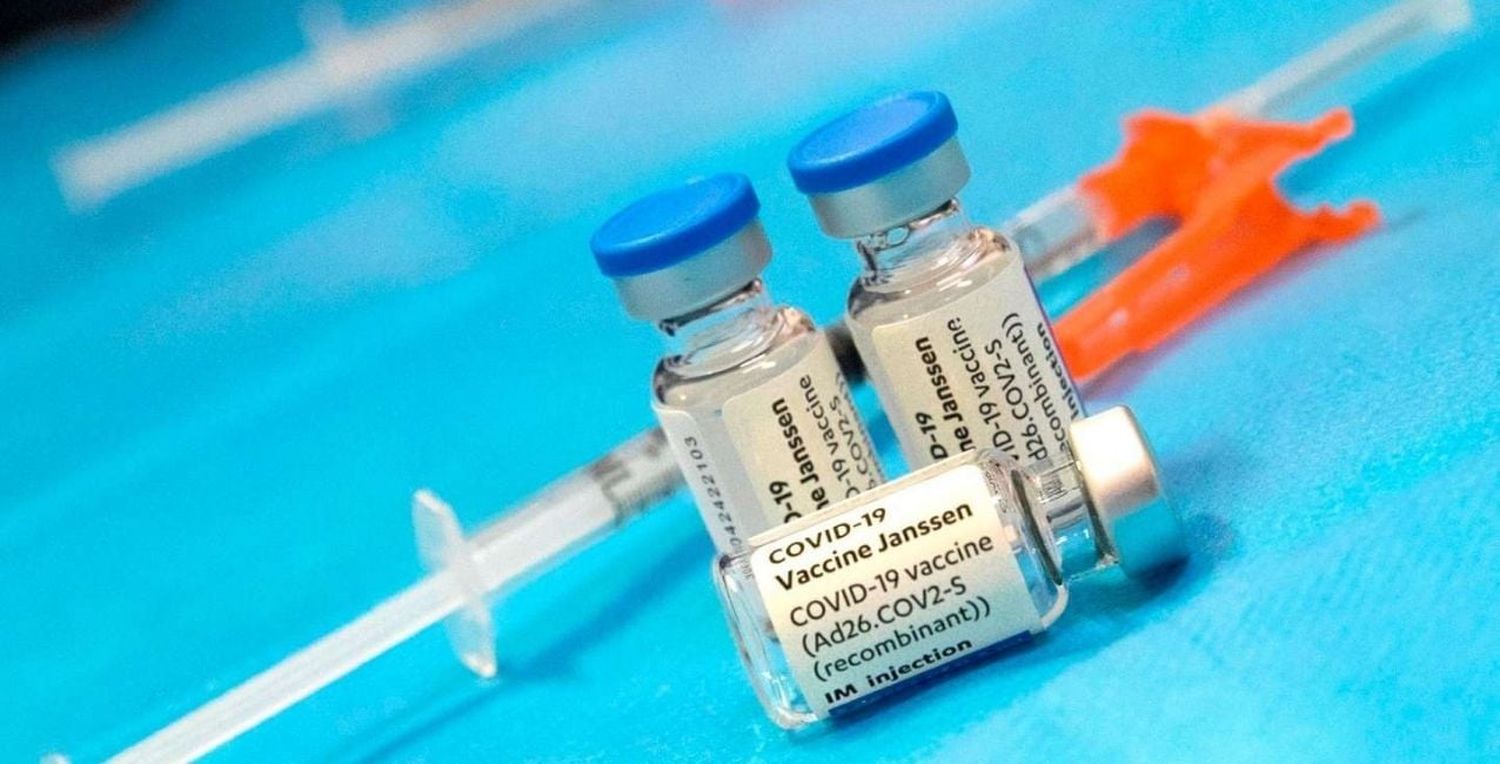
El trámite lo había iniciado el laboratorio Johnson & Johnson en diciembre, pero recién en junio había sumado nueva documentación. El Gobierno esperaba su aprobación para poder comprarla, pero también para recibir donaciones de Estados Unidos.
La vacuna Janssen contra el coronavirus, del laboratorio estadounidense Johnson & Johnson, recibió la aprobación de emergencia por parte de la Administración Nacional de Medicamentos, Alimentos y Tecnología Médica (ANMAT), un paso fundamental que el Gobierno esperaba para avanzar en la compra de esos sueros monodosis, pero también para poder recibir donaciones de la Administración de Joe Biden.
La autoridad regulatoria dio el visto bueno el viernes, pero la información recién trascendió este martes y ya fue publicada en su página web.
La Casa Rosada intenta cortar la circulación del virus antes de que termine el 2021. El porcentaje de inoculaciones que se debe alcanzar y cómo sigue la polémica por el uso de las dosis de Sinopharm en los niños.
Coronavirus: aumentaron “levemente” los casos de variante Delta en Argentina
Por tratarse de un pedido realizado por el laboratorio, es la ANMAT la que debe autorizar de emergencia la vacuna, tal como lo hizo con la de Pfizer, por ejemplo.
Johnson & Johnson había iniciado el trámite en diciembre del año pasado, pero recién en junio presentó nuevos documentos que se sumaron al expediente y que terminaron ahora con la aprobación de emergencia.
En la última semana de junio, el Gobierno modificó por decreto la Ley de Vacunas y quitó la palabra negligencia de la normativa -además de otros cambios- para garantizar un marco legal acorde a los requerimientos de estos laboratorios extranjeros. Gracias a ese procedimiento, ya ingresaron al país vacunas de Pfizer y de Moderna.
Qué dice la autorización de emergencia de la ANMAT para la vacuna de Janssen
En la disposición 7502, la ANMAT indica que “e virtud de tratarse de una autorización condicional, la vigencia del Certificado será de un año” y que en forma previa el laboratorio deberá solicitar “la autorización efectiva de comercialización notificando fecha de inicio de la importación del primer lote a comercializar a los fines de realizar la verificación técnica correspondiente”.
Además, ordena que el laboratorio “deberá presentar los informes de avance, las modificaciones y las actualizaciones del Plan de Gestión de Riesgo ante el INAME (Instituto Nacional de Medicamentos)” y que también “deberá presentar los informes de seguridad periódicos cada seis meses luego de la comercialización efectiva del producto ante el INAME”.
Lo confirmó la ANAC a través de un comunicado. La medida se basa en haber alcanzado el umbral del 50% de la población vacunada con esquema completo. Ese día, quienes tengan doble dosis quedarán eximidos de la presentación del test de antígenos.
Autorizan la aplicación de la vacuna Moderna para mayores de 12 años
Otro de los puntos que debe tenerse en cuenta es que “todo cambio en el perfil de seguridad o de eficacia del producto deberá evidenciarse en la correspondiente modificación del prospecto”; a la vez que advierte que “en caso de incumplimiento de las obligaciones previstas en los artículos precedentes” la ANMAT “podrá suspender la comercialización del producto” siempre que “consideraciones de salud pública así lo ameriten”.
El viernes pasado, la Agencia Europea de Medicamentos (EMA) concluyó que existe “un posible vínculo” entre varios casos raros de tromboembolismo venoso (TEV) y la vacuna de Janssen, por lo que incluyó esta afección en el prospecto como un efecto secundario poco común.